Regulation (EU) 2017/745- clinical trials application process

After almost 25 years, the law on medical devices has changed. The “old” EU Directives (Medical Device Directive (MDD) 93/42 / EEC and Active Implantable Medical Devices Directive (AIMDD) 90/385 / EEC) were replaced by the Medical Device Regulation (MDR) 2017/475. European Union law considering medical devices has changed from directives to the regulation, which means that now the medical devices’ EU law is directly applicable at national level without requiring transposition through specific national legislation. This leads to the harmonization of rules on medical devices across the EU. According to MDR 2017/475 the aim of this regulation is “to ensure the smooth functioning of the internal market as regards medical devices, taking as a base a high level of protection of health for patients and users, and taking into account the small- and medium-sized enterprises that are active in this sector. At the same time, this Regulation sets high standards of quality and safety for medical devices in order to meet common safety concerns as regards such products.” The MDR applies since 26 May 2021, following a four-year transition period.
Changes to the European law on medical devices also applied to clinical investigation. Devices are classified into classes I, IIa, IIb and III, taking into account the intended use of the medical devices and the associated risks. Before being placed on the market, not every medical device has to be subject to clinical investigation. Only in the case of class III devices and implantable devices such a clinical investigation must be performed, although there are some exceptions in this regard.
According to MDR 2017/475, there are 3 possible regulatory pathways for submission of application for initiation of a clinical investigations of a medical device, it all depends on the current status (if it has CE mark) of the device and the future use of the clinical investigation results:
- Art. 62.* for medical device not yet CE mark, the results of the clinical investigation may be used for conformity procedure to get CE mark, or* for medical device CE mark, the clinical investigation will not be conducted within intended purpose but the results of the study may be used for conformity procedure to get CE mark.
- Art. 74 (1) – for medical device CE mark, the clinical investigation is to be conducted to further assess, within the scope of its intended purpose, and the investigation would involve submitting subjects to procedures additional to those performed under the normal conditions of use of the device and those additional procedures are invasive or burdensome
- Art. 82 – other clinical investigations (e.g. non-interventional study)
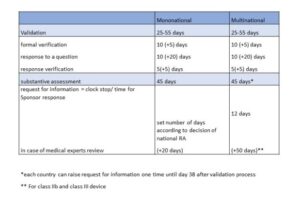
Below are presented timelines for the regulatory pathway according to the Art. 62 of MDR, which describes process of evaluation of application for initiation of a clinical investigations by Regulatory Authority (RA). An opinion of the Ethics Committee (EC) is also required, but the timing of the request for an opinion is governed by local law. Starting a clinical trial is possible when:
- for investigational medical devices of class I or for non-invasive devices of class IIa and class IIb (unless national law provides otherwise) – immediately after validation of the application by the RA and when a positive opinion of the EC is issued, valid in a given country, in accordance with national law
- for investigational medical devices other than those referred to in point (a) – immediately after the Member State concerned notifies the Sponsor that the authorization has been granted and provided the positive opinion of the EC is valid for the entire Member State in accordance with national law
According to Article 62, it is possible to make application for clinical investigation in Member State (mononational) or in many Member States simultaneously (multinational). This is to be made possible through the appropriate functionality of the special EUDAMED medical device database. However, at present, for the applicant, this database is not yet fully functional, and applications must be submitted directly to the RA, making simultaneous submission in multiple Member States impossible for the time being.
In the case of an application for a clinical investigation in accordance with Article 74, the Sponsor should notify the concerned Member States at least 30 days prior to its commencement via the EUDAMED electronic system. Currently, due to the lack of full functionality of the system, such applications should be submitted directly to the RA. An opinion of the Ethics Committee (EC) is also required, but the timing of the request for an opinion is governed by local law.
As regards the submission of an application for a clinical trial under Art. 82, it is to be governed by local law. Applying for clinical investigation under MDR is not yet a fully regulated process. Some of the activity is regulated by MDR and some by local law, which in some Member States still has not been fully established. The lack of a fully functional EUDAMED database also means that RAs from different Member States have to use temporary solutions for certain procedures that were to be handled electronically by the EUDAMED database.
In the case of problems related to the application process or the preparation of appropriate documentation necessary for the application for a clinical trial, Clinmark provides assistance at every stage of the process. Please contact our team to ask for details about our services and possibilities.